Why our DNA accumulates errors with age, how those errors ripple through cells and tissues, and what current research suggests we can do about it.
TL;DR
- Somatic DNA changes accumulate with age across many human tissues. Different cell types acquire distinct patterns of damage. Some of these changes reshape tissue function long before disease appears. Cell+1
- Mitochondrial DNA damage and leakage stoke inflammation through the cGAS–STING pathway, tying broken genomes to “inflammaging.” Blocking this pathway in mice reduces age-related inflammation and improves tissue function. Nature+1
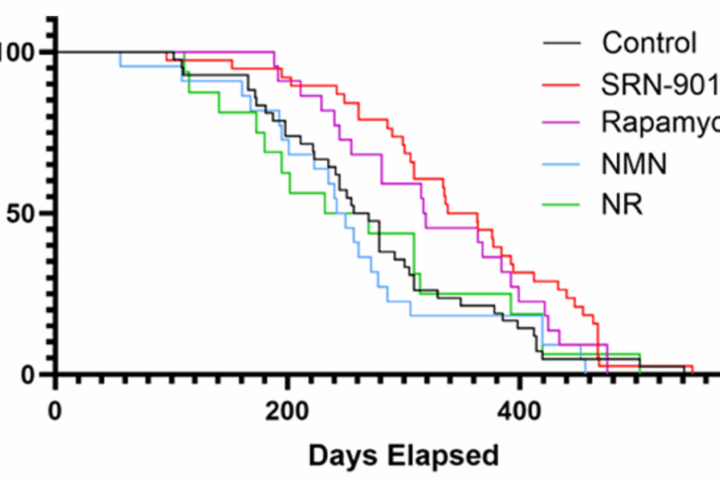
- Telomere dysfunction is a potent driver of genomic instability and cellular senescence. In new mouse studies, carefully controlled telomerase activation extends lifespan without obvious cancer signals. Nature+2PubMed+2
- Clonal hematopoiesis provides a living example of age-linked genome change that alters disease risk, including atherosclerosis. Nature+1
- Interventions that reduce DNA damage or mute its inflammatory sequelae are emerging. Dietary restriction can dampen cGAS–STING signaling in DNA-repair-deficient mice, and exercise shows early signs of protecting genomes in older adults. Evidence in humans is still developing. PubMed+2
What “genomic instability” means, in plain English
Every cell carries a copy of your genome. Over time, chemical insults, replication mishaps, and simple chance introduce errors. Instability is the rising tide of those errors and the failures to repair them. It includes single-letter substitutions, insertions and deletions, large structural rearrangements, mitochondrial genome lesions, transposon reactivations, and chromosome mis-segregation events such as aneuploidy. The concept sits at the core of modern aging biology and interlocks with other hallmarks like telomere attrition, epigenetic drift, and senescence. Nature
A map of age-linked mutations across human tissues
Thanks to single-cell and low-artifact sequencing, researchers can now watch mutations accumulate in specific cell types. In the human brain, neurons and oligodendrocytes age under distinct mutational “weathers,” implying different damage sources and repair bottlenecks. Oligodendrocytes show processes that diverge from neurons as years accrue. Cell
A 2025 multi-omic brain study reinforced the theme: somatic mutations in neurons increase with age and correlate with transcriptional changes that may undermine circuit function. Nature
These cell-type-specific patterns sit atop a broader rule observed across mammals. Somatic mutation rates generally scale with species lifespan, suggesting evolution tunes damage and repair to life history. Nature
Mitochondrial genomes: small circles with big consequences
Mitochondrial DNA (mtDNA) accrues point mutations and deletions with age. New work shows extensive mtDNA mosaicism in normal human cells, with thousands of variants cataloged across donors and tissues. These variants can clonally expand and, in certain contexts, impair bioenergetics. Nature
When damaged mitochondrial DNA escapes into the cytosol, it triggers the cGAS–STING innate immune pathway. In older mice, cytosolic mtDNA drives type I interferon responses and chronic inflammation in retina, a striking example of organ-specific inflammaging seeded by genome fragments. Nature
Reviews in 2024–2025 synthesize the case that mtDNA mutations and mitochondrial dysfunction are tightly interwoven with organismal aging. Frontiers+1
The fuse at chromosome ends: telomeres and lifespan
Telomeres shorten with each cell division and are hypersensitive to oxidative damage. Their dysfunction activates persistent DNA damage signaling and pushes cells into senescence. A comprehensive 2022 review outlines how telomere damage contributes to age-related pathologies, from marrow failure to pulmonary fibrosis. Nature
Safety had long been the barrier to therapeutic telomerase activation. That is shifting. A 2024 knock-in mouse study that placed Tert under a ubiquitous but controlled promoter extended lifespan, lengthened telomeres, and did not reveal spontaneous genotoxicity within the study window. A 2025 follow-up refined telomerase gene control and corroborated lifespan effects in telomerase-deficient lines. PubMed+1
Beyond lengthening telomeres, TERT appears to remodel the epigenome. A 2024 study in Cell reported that TERT activation influences DNA methylation programs that track with biological aging, hinting that telomerase may stabilize genome function through multiple routes. Cell
DNA repair: the quiet machinery that ages with us
Cells deploy several repair systems. Nonhomologous end joining and homologous recombination seal double-strand breaks. Base excision repair cleans up oxidation scars. Nucleotide excision repair removes bulky lesions. With age, capacity and coordination decline in many contexts. In human oocytes, researchers observed reduced mobility of DNA damage sites and cohesin loss that lowers repair efficiency, providing a mechanistic link to maternal-age aneuploidy. Cell
Recent reviews aggregate cross-species data suggesting that lifetime changes in repair pathway choice and regulation shape how damage accumulates and how cells respond to it. ScienceDirect
When jumping genes wake up
LINE-1 retrotransposons are genomic hitchhikers that can copy and paste themselves, potentially disrupting genes and igniting inflammation. Multiple 2024 papers sharpened the mechanism: the transcription factor PAX5 can activate LINE-1 and drive senescence, while SIRT6 counteracts this activation. New small molecules that target LINE-1 machinery are under exploration. PMC+1
In mammalian brains, LINE-1 proteins rise with age. Although causality in humans is not settled, these observations align with animal studies and bolster a model where retroelements contribute to neuroinflammation late in life. PMC
The cGAS–STING link between broken DNA and chronic inflammation
Cells treat misplaced DNA as a danger signal. The cGAS enzyme senses cytosolic DNA and activates STING, which turns on interferons and other inflammatory genes. In 2023, investigators demonstrated that blocking STING in aged mice reduced inflammatory phenotypes in multiple organs and improved function. In the brain, STING activation drove reactive microglial states and cognitive decline. Nature
Fresh data in 2024–2025 paint a nuanced picture. Mitochondrial DNA release with senescence can tip cGAS–STING toward pathologic immune states, yet complete loss of cGAS in mice shortened lifespan and unleashed retroelements. Context, dose, and timing matter. Cell+1
A natural human experiment: clonal hematopoiesis
With age, single blood stem cells that acquire driver mutations can expand, a phenomenon called clonal hematopoiesis. Carriers have elevated risks of blood cancers and vascular disease. A 2024 longitudinal study combined deep sequencing and vascular imaging and found that individuals with clonal hematopoiesis had higher odds of developing femoral atherosclerosis over six years, while atherosclerosis did not appear to drive clone expansion over that interval. Nature
This is genome instability changing disease risk at population scale, often decades before malignancy. Nature
Nuclear architecture and progeroid clues
Premature aging disorders highlight genome maintenance as a root cause. In Hutchinson–Gilford progeria, mutant lamin A destabilizes the nuclear envelope and promotes persistent DNA damage. In Werner syndrome, loss of the WRN helicase compromises replication and repair. These pathologies accelerate many features of normal aging and continue to inform targets such as WRN itself, which has become a synthetic-lethal vulnerability in microsatellite-instable cancers. PLOS+2PMC+2
Concrete results that move the field
- Cell-type mutation signatures with age. Human oligodendrocytes and neurons accumulate distinct somatic mutations over time, implying different damage sources and repair capacities inside the same brain. Cell
- mtDNA-to-inflammation conduit. Old mouse retina shows cytosolic mtDNA that activates cGAS–STING and type I interferon, positioning mitochondrial genome leakage as a proximate trigger of inflammaging in a living tissue. Nature
- STING blockade improves aged tissues. In aged mice, pharmacologic STING inhibition reduced inflammatory markers across organs and improved function, while in the brain STING activation alone drove neurodegeneration signatures. Nature
- Telomerase as a longevity lever in vivo. Knock-in mice that upregulate Tert showed elongated telomeres, better tissue repair, and lifespan extension without apparent genotoxicity in the tested windows. PubMed+1
- Dietary restriction tames a hyper-unstable genome. In DNA-repair-deficient Ercc1Δ/− mice, dietary restriction mitigated vascular aging and modulated cGAS–STING signaling, pointing to systemic levers that operate upstream of repair bottlenecks. PMC+1
- Clonal hematopoiesis predicts new atherosclerosis. In middle-aged adults without baseline disease, detectable driver mutations in blood stem cells increased the risk of developing femoral artery plaques over six years. Nature
What is actionable today, and what remains speculative
Lifestyle levers
Small but growing bodies of evidence suggest that structured aerobic exercise in later life can reduce markers of genomic instability, including telomere dysfunction, in humans. These studies are early and often presented first at conferences, yet they align with decades of mechanistic work linking physical activity to improved DNA repair dynamics and lower oxidative burden. Medical Xpress+1
Dietary restriction and protein modulation show robust effects in short-lived models. In DNA-repair-deficient mice, restriction improved vascular aging and dialed down cGAS–STING signaling, consistent with the idea that lowering damage load reduces inflammatory amplification. Translation to ad libitum humans is not settled. PubMed
Pharmacology and advanced modalities
NAD+ augmentation is being tested as a way to support PARP-dependent DNA repair and sirtuin functions. Trials reliably raise blood NAD+, but durable functional benefits remain to be proven in large, long-duration cohorts. PMC+1
Targeting retrotransposons with reverse transcriptase inhibitors has reduced inflammation and rescued cognition in multiple mouse contexts. Whether this generalizes to aging humans is unknown, but the mechanistic rationale continues to strengthen. Frontiers
cGAS–STING inhibitors are moving from cancer and autoimmunity into aging models. Early mouse work shows improved progeroid phenotypes with a small-molecule STING inhibitor, while complete genetic ablation of cGAS can shorten lifespan and de-repress retroelements. Dosing and timing will be critical. PMC+1
Telomerase activation is crossing an inflection point in preclinical systems. Mouse studies report lifespan extension without obvious tumor acceleration under controlled expression. How to translate that safety profile to humans is the central question. PubMed
How genomic instability explains so many age-linked disorders
- Neurodegeneration. Distinct mutation spectra in glia and neurons, rising LINE-1 expression, and sustained cGAS–STING activation provide multiple routes to synaptic failure and cognitive decline with age. Cell+2PMC+2
- Cardiometabolic disease. mtDNA damage provokes sterile inflammation in metabolically active tissues. In the vasculature, clonal hematopoiesis changes immune programming and increases atherosclerosis risk. Nature+1
- Reproductive aging. Age-linked loss of cohesin and slower DNA repair in oocytes increase aneuploidy risk, contributing to infertility and adverse outcomes in later maternal age. Cell
Where the field is headed
- Cell-type atlases of damage. Expect richer maps that quantify mutations, repair kinetics, and inflammatory tone in the same cells, across ages and tissues. Nature
- Precision control of genome guardians. From pharmacologic p53 tuning that reduces cytoplasmic chromatin fragments and inflammatory signaling in aged tissues, to selective STING modulation that avoids immune compromise. Nature
- Genome housekeeping for mitochondria. Selective editing or removal of deleterious mtDNA species and reinforcement of mitophagy to prevent mtDNA leakage. Nature+1
- Taming transposons. Small molecules and epigenetic strategies that keep LINE-1 quiet without collateral damage. Nature
- Cautious rejuvenation. Partial reprogramming and telomerase gene control tested with better safety circuits and tissue targeting. Liebert Publishing+1
Bottom line
Genomic instability is not a single lesion. It is a network of injuries and imperfect repairs that amplifies with age and connects directly to inflammation, senescence, and tissue failure. In the last two years, the field moved from metaphors to mechanisms in living tissues: mapping somatic mutations cell by cell, tracing mtDNA to cGAS–STING, and showing that carefully tuned telomerase and innate immune modulators can shift aging phenotypes in mammals. The translation challenge now is to preserve protective DNA sensing and repair while silencing their chronic, damaging echoes.
References
- López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243–278. https://doi.org/10.1016/j.cell.2022.11.001
- Cagan, A., Baez-Ortega, A., Brzozowska, N., Abascal, F., Coorens, T. H. H., … Martincorena, I. (2022). Somatic mutation rates scale with lifespan across mammals. Nature, 604, 517–524. https://doi.org/10.1038/s41586-022-04618-z
- Ganz, J., Luquette, L. J., et al. (2024). Contrasting somatic mutation patterns in aging human neurons and oligodendrocytes. Cell, 187(8), 1955–1970.e1923. https://doi.org/10.1016/j.cell.2024.02.025
- Gulen, M. F., Samson, N., Keller, A., Schwabenland, M., Liu, C., Glück, S., et al. (2023). cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature, 620, 374–380. https://doi.org/10.1038/s41586-023-06373-1
- Vermeij, W. P., Dollé, M. E. T., Reiling, E., Jaarsma, D., Payán-Gómez, C., Bombardieri, C. R., et al. (2016). Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature, 537(7620), 427–431. https://doi.org/10.1038/nature19329
- Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., et al. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. The New England Journal of Medicine, 377(2), 111–121. https://doi.org/10.1056/NEJMoa1701719
- Fuster, J. J., MacLauchlan, S., Zuriaga, M. A., Polackal, M. N., Ostriker, A. C., Chakraborty, R., et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science, 355(6327), 842–847. https://doi.org/10.1126/science.aag1381
- D’Ordine, R. L., Xie, Y., Byrne, K., et al. (2024). Structure-guided high-throughput discovery of the first drug-like LINE-1 endonuclease inhibitors. Nature Communications. https://doi.org/10.1038/s41467-024-50745-8
- Sharma, R., et al. (2024). DNA damage repair coordinates age-associated oocyte aneuploidy. Current Biology. https://doi.org/10.1016/j.cub.2024.05.012
- Mihalas, B. P., et al. (2024). Reversal of age-related cohesion loss increases mouse oocyte viability. Current Biology. https://doi.org/10.1016/j.cub.2024.04.004
- Huang, Y., et al. (2023). Mechanism and therapeutic potential of targeting cGAS–STING in neurodegenerative diseases. Molecular Neurodegeneration, 18, 70. https://doi.org/10.1186/s13024-023-00672-x
- Brito de Araújo, M., Egger, F., et al. (2025). Physical activity, exercise and telomere length: An umbrella review of systematic reviews and meta-analyses. British Journal of Sports Medicine. https://doi.org/10.1136/bjsports-2024-110431
- Victorelli, S., Muñoz-Espín, D., & Gil, J. (2022). The burden of telomere dysfunction in age-related diseases. Nature Reviews Genetics, 23, 425–442. https://doi.org/10.1038/s41576-022-00532-5
- Capasso, M., et al. (2024). Single-cell whole-genome sequencing of brain cells: Age, disease and technical considerations. Signal Transduction and Targeted Therapy, 9, 296. (Cites Ganz et al., 2024, Cell.) https://pmc.ncbi.nlm.nih.gov/articles/PMC11310189/