Main Points
- A new study proposes ferro-aging, a mechanism linking iron buildup to cellular damage and senescence
- The enzyme ACSL4 emerges as a central driver of lipid vulnerability and oxidative stress
- Long-term primate data suggest vitamin C modulates this pathway, but not in the way most people assume
- The work reframes aging as partly a problem of metal and membrane chemistry, not just genes or inflammation
Aging is often described as a slow accumulation of damage. But what if part of that damage isn’t random at all? What if it follows a specific chemical script, written in iron and lipids, quietly reshaping our cells over decades?
A recent paper in Cell Metabolism proposes exactly that. It introduces a term that sounds almost too neat for biology: ferro-aging. Not a metaphor, but a mechanism. Not just rust, but regulated decay.
And buried within that mechanism is an unexpected player. Not a new drug, not a gene-editing tool, but something far more familiar.
Why this matters
Most longevity strategies today cluster around a few dominant ideas. Remove senescent cells. Reprogram epigenetics. Improve mitochondrial function. Adjust metabolism. Each of these approaches targets a different layer of the aging process, often without a unifying thread.
This paper attempts to connect several of those layers through a single axis: iron-driven lipid damage leading to cellular senescence and tissue dysfunction. If correct, it suggests that aging isn’t just about accumulating damage, but about entering a specific biochemical state that can, at least partially, be modulated.
That shift matters because it reframes intervention. Instead of asking how to repair aging after it occurs, it asks whether we can interrupt the chemistry that creates it.
The quiet chemistry behind aging
Aging biology has increasingly converged on a set of recurring themes, often referred to as the hallmarks of aging. Among them, cellular senescence stands out. Senescent cells are those that stop dividing but refuse to die, secreting inflammatory factors that disrupt surrounding tissue.
But senescence does not arise in isolation. It is triggered by stress, and one of the most underappreciated sources of that stress is lipid peroxidation. This process occurs when reactive molecules attack the fatty components of cell membranes, particularly polyunsaturated fatty acids, which are structurally vulnerable.
Iron plays a catalytic role in this process. It accelerates the formation of reactive oxygen species, which in turn attack lipids. Over time, this creates a feedback loop of damage, inflammation, and dysfunction.
The novelty here is not that iron or lipid peroxidation are involved in aging. That has been known for decades. The novelty is the proposal that these processes form a coordinated, regulated axis, rather than a collection of downstream effects.
The authors call this axis ferro-aging.
Designing an experiment that spans species and time
The study is ambitious in scope. It moves across systems, from cultured human cells to aged mice to non-human primates. This breadth is not decorative. It is necessary to argue that ferro-aging is not an artifact of a single model.
In cellular systems, the researchers induced iron accumulation using controlled treatments. They then tracked markers of lipid peroxidation, senescence, and metabolic dysfunction. These experiments allowed them to establish causality in a controlled environment.
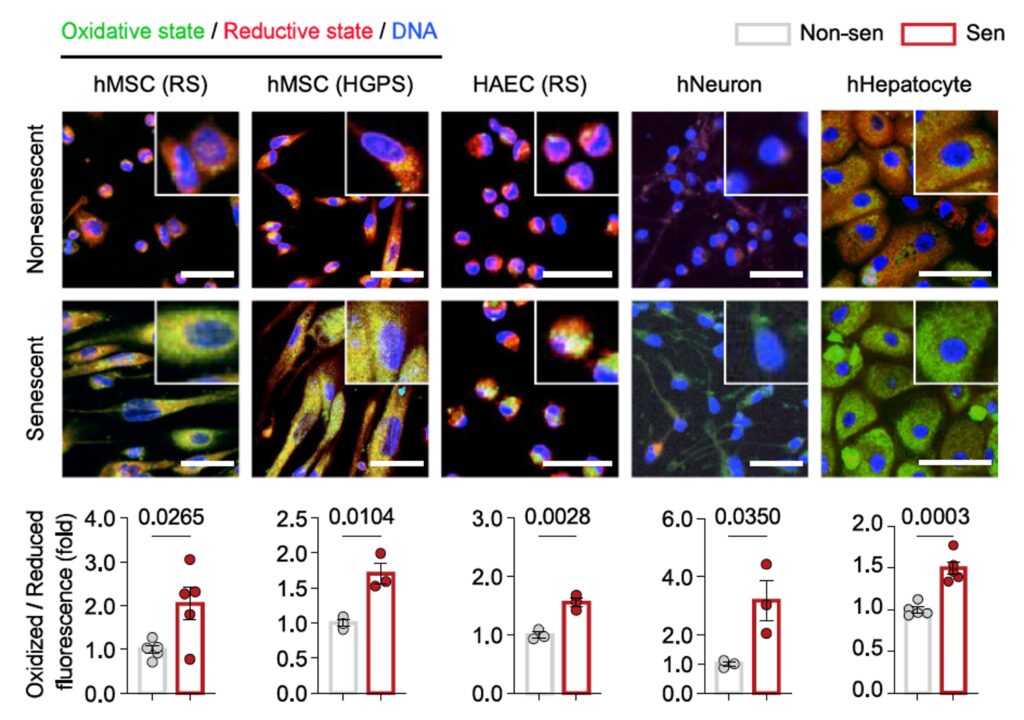
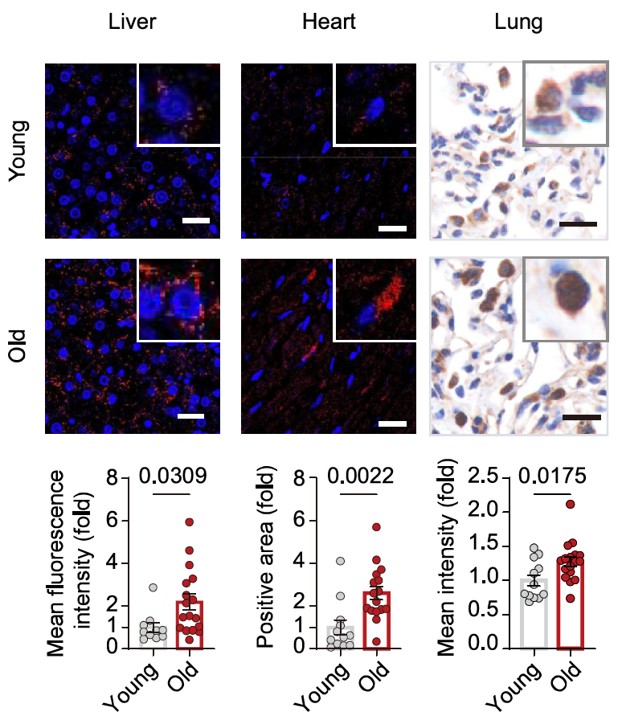
In parallel, they examined aged tissues. Human samples, elderly immune cells, and primate tissues all showed consistent signatures: increased iron, increased lipid damage, and increased expression of specific metabolic enzymes.
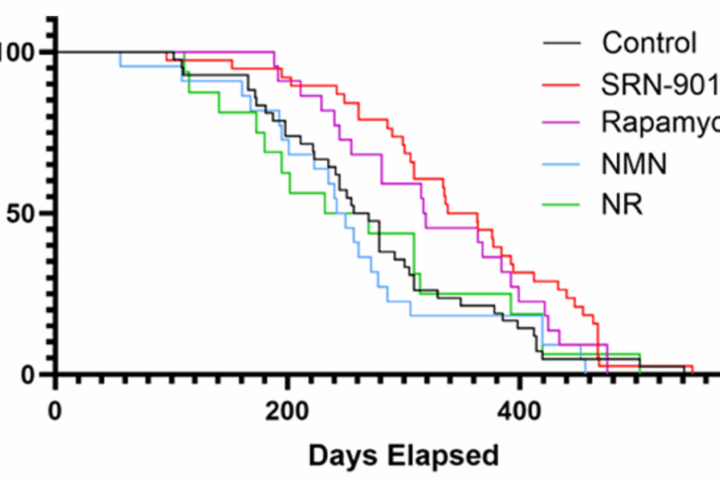
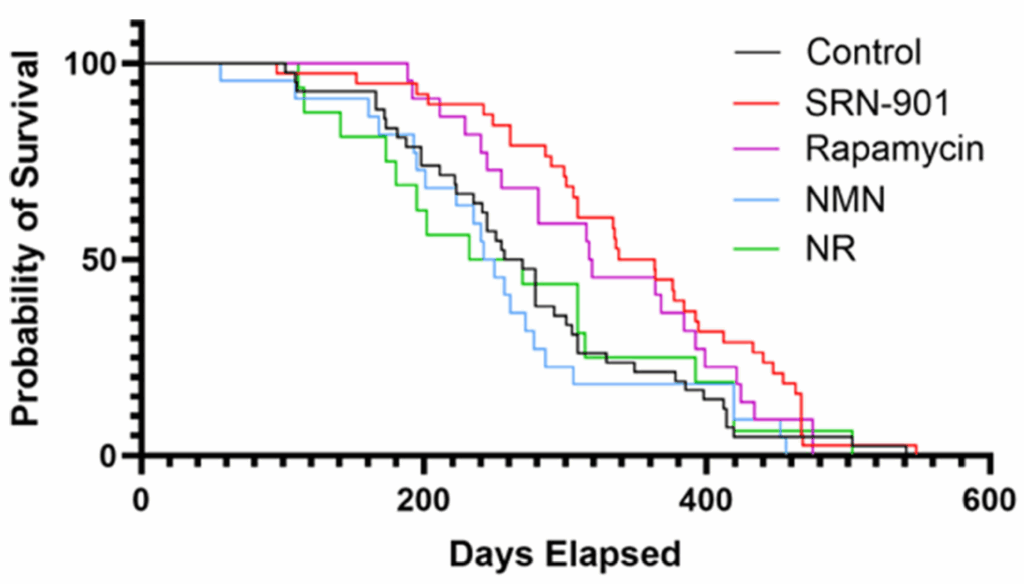
The study then moves into intervention. In mice, they manipulated a key enzyme and observed downstream effects on physiology and behavior. Finally, in one of the more unusual aspects of the paper, they conducted a 40-month intervention in cynomolgus monkeys, measuring structural, metabolic, and molecular changes over time.
This progression from reductionist to organismal to primate is rare. It doesn’t eliminate uncertainty, but it significantly raises the bar for relevance.
A single enzyme at the center of the storm
At the core of the ferro-aging hypothesis is ACSL4. This enzyme helps incorporate long-chain fatty acids into cellular membranes. On the surface, this is a mundane metabolic function.
But not all lipids are equal. ACSL4 preferentially enriches membranes with fatty acids that are particularly susceptible to oxidation. In effect, it determines how flammable the membrane is in the presence of oxidative stress.
The data show that ACSL4 increases with age across multiple systems. When ACSL4 is overexpressed in cells, lipid peroxidation rises, and senescence markers follow. When ACSL4 is suppressed, the opposite occurs.
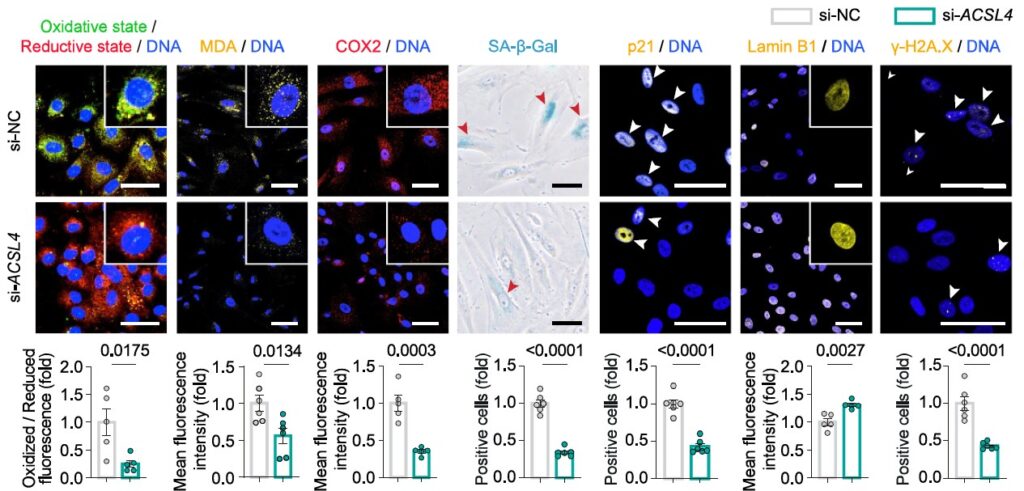
In mice, targeted suppression of ACSL4 in the liver led to improvements in multiple domains. Cognitive function improved. Motor coordination improved. Biomarkers of senescence declined.
This is where the paper becomes mechanistically compelling. It’s not simply correlating iron and aging. It’s identifying a control point that links iron metabolism, membrane composition, and cellular fate.
What they found, in concrete terms
Across systems, the findings converge on a consistent pattern.
Iron levels increase with age, particularly in a reactive form. Lipid peroxidation markers, such as malondialdehyde, increase in parallel. ACSL4 expression rises, amplifying membrane susceptibility to oxidative damage.
In cellular models, increasing iron leads to increased ACSL4, increased lipid damage, and increased senescence markers. Reducing ACSL4 interrupts this cascade.
In mice fed high-iron diets, functional decline emerges rapidly. Cognitive performance drops. Muscle strength decreases. Coordination worsens. These are not subtle shifts.
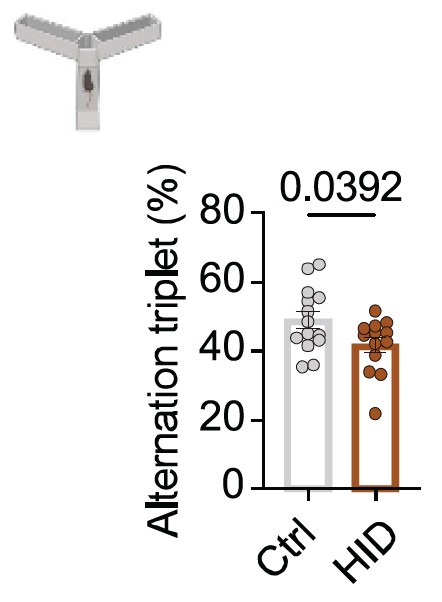
In primates, long-term intervention reduced lipid peroxidation, improved metabolic markers, and attenuated brain atrophy as measured by imaging. These outcomes are directionally consistent with slowed aging processes, though they stop short of demonstrating lifespan extension.
Importantly, fibrosis and certain advanced pathologies were not significantly reversed. This suggests limits to the intervention, especially in later-stage disease.
The vitamin C result, stripped of mythology
The most attention-grabbing aspect of the paper is the identification of vitamin C as an inhibitor of ACSL4 activity in their system.
It’s tempting to interpret this through the lens of antioxidants. That would be a mistake. The data suggest a more specific interaction, where vitamin C modulates a pathway upstream of lipid peroxidation rather than simply scavenging free radicals.
In cell systems, vitamin C reduced lipid damage and partially restored cellular function. In primates, long-term administration reduced ferro-aging signatures across multiple tissues.
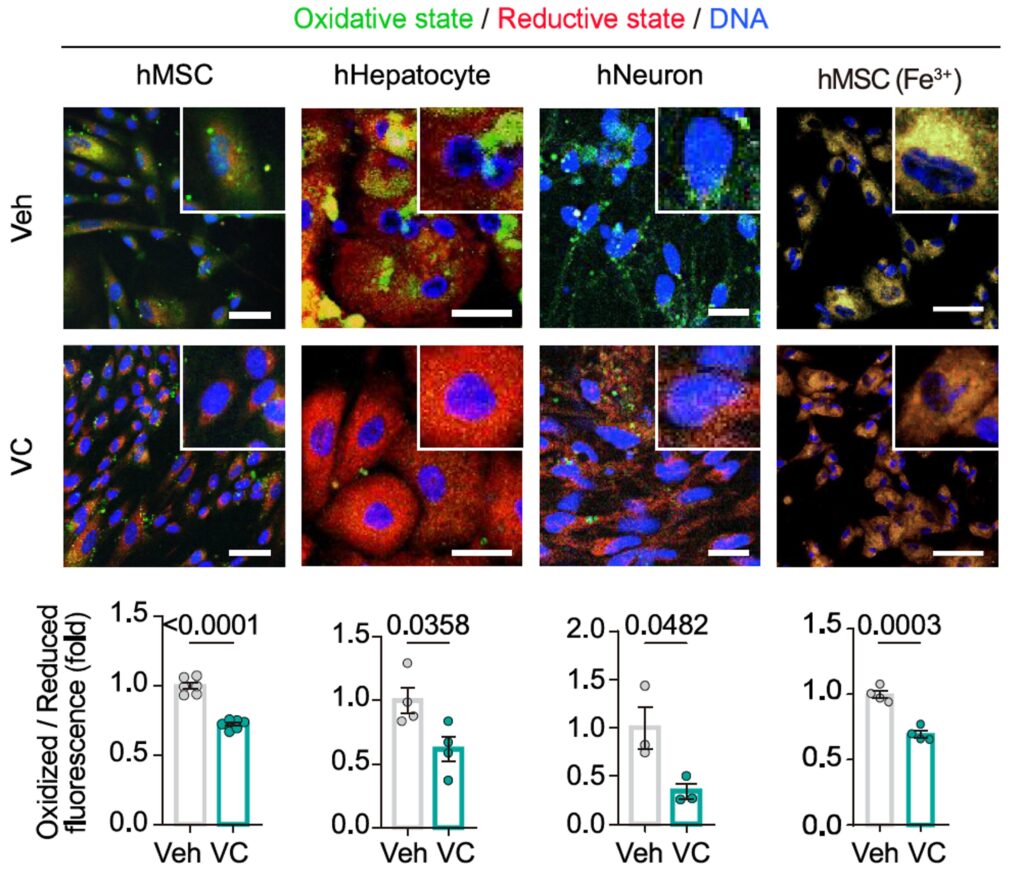
But this is where caution is essential. Vitamin C is a pleiotropic molecule. It affects redox balance, collagen synthesis, immune signaling, and more. The study cannot fully isolate ACSL4 inhibition from these broader effects.
The signal is real. The mechanism is plausible. The specificity is not yet proven.
Aging as a membrane problem
If the ferro-aging model holds, it reframes aging in a subtle but important way.
Instead of viewing aging primarily as a failure of DNA, proteins, or mitochondria, it suggests that cell membranes themselves are a central battleground. Their composition determines vulnerability. Their oxidation drives dysfunction.
This perspective integrates several hallmarks. Lipid peroxidation contributes to senescence. Senescence drives inflammation. Inflammation disrupts metabolism. Metabolic dysfunction feeds back into oxidative stress.
The model is appealing because it is both chemical and systemic. It doesn’t replace existing frameworks. It connects them.
Where the story remains incomplete
Despite its strengths, the study leaves critical questions unresolved.
First, causality across tissues remains uneven. The strongest mechanistic data come from liver-focused interventions. Whether the same ACSL4-driven process dominates in brain, muscle, or vasculature is less clear.
Second, the primate data, while compelling, are not definitive. Improvements in biomarkers and imaging do not necessarily translate to extended lifespan or delayed disease onset. The study is long for a primate experiment, but still short relative to aging itself.
Third, the vitamin C finding risks overextension. Without precise dose-response mapping, tissue specificity, and mechanistic isolation, it is premature to treat it as a targeted therapeutic.
Finally, ferro-aging itself may not be a single pathway. It may be a convergence of multiple stress responses that appear unified under certain conditions.
What this means for longevity science
The most durable contribution of this work is not vitamin C. It is the identification of ACSL4-mediated lipid vulnerability as a potential lever in aging biology.
For senolytics, this suggests an upstream strategy. Instead of clearing senescent cells, prevent their formation by stabilizing membranes and reducing oxidative triggers.
For metabolic interventions, it reinforces the importance of lipid composition, not just lipid quantity. The types of fats incorporated into membranes may matter as much as overall metabolic health.
For translational work, the primate data provide a rare bridge between rodent models and human relevance. Not proof, but a signal worth following.
And for the field more broadly, it adds a layer of specificity. Aging may not just be accumulation. It may be entry into distinct biochemical states, each with its own vulnerabilities and intervention points.
Sources
- Liu, L., Zheng, Z., You, W., Yang, P., Wen, Y., Qiao, Y., Ma, S., Zhang, H., Zhang, S., Xu, G., Ma, C., Tian, A., Jiang, M., Zhang, T., Geng, L., Li, J., Sun, X., Wang, F., Xiong, M., … Liu, G.-H. (2026). Vitamin C inhibits ACSL4 to alleviate ferro-aging in primates. Cell Metabolism, 38(4), 673–693.e17.
- López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243–278.
- López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217.
- Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., Patel, D. N., Bauer, A. J., Cantley, A. M., Yang, W. S., Morrison, B., III, & Stockwell, B. R. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell, 149(5), 1060–1072.
- Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., Irmler, M., Beckers, J., Aichler, M., Walch, A., Prokisch, H., Trümbach, D., Mao, G., Qu, F., Bayir, H., Füllekrug, J., Scheel, C. H., Wurst, W., Schick, J. A., … Conrad, M. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology, 13(1), 91–98.
- Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., & Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences, 113(34), E4966–E4975.
- Hentze, M. W., Muckenthaler, M. U., & Andrews, N. C. (2004). Balancing acts: Molecular control of mammalian iron metabolism. Cell, 117(3), 285–297.
- Minotti, G., & Aust, S. D. (1989). The role of iron in oxygen radical mediated lipid peroxidation. Chemico-Biological Interactions, 71(1), 1–19.
- Campisi, J., & d’Adda di Fagagna, F. (2007). Cellular senescence: When bad things happen to good cells. Nature Reviews Molecular Cell Biology, 8(9), 729–740.