Main Points:
- A distinct p21+TREM2+ senescent macrophage state is identified, separate from classical immune activation
- These cells amplify inflammaging via mtDNA–cGAS–STING–type I interferon signaling
- They accumulate in aging liver and metabolic disease, impairing lipid handling and tissue repair
- Senolytic clearance reduces inflammation and steatosis in mice, but translation remains uncertain
For years, aging biology has had a convenient villain: the senescent cell. It lingers, refuses to die, and poisons its surroundings. But the story has always been a little too clean. Not all cells fit neatly into that role, and some, like macrophages, were thought to be too dynamic, too adaptive, to truly become senescent.
A new paper in Nature Aging complicates that narrative in a useful way. It suggests that one of the immune system’s most flexible cells can, under the right pressures, lock into a dysfunctional, inflammatory state that looks and behaves like senescence. And once there, it doesn’t just contribute to aging. It may help drive it.
When the Cleanup Crew Gets it Wrong
Aging is often described as a gradual accumulation of damage, but the real problem isn’t just the damage itself. It’s how the body responds to it. Over time, repair systems become distorted, inflammatory signals persist, and cells that should clear debris begin to perpetuate it.
This study reframes part of that failure. Instead of viewing macrophages as responders to aging-related damage, it positions a subset of them as participants in the pathology, locked into a self-sustaining inflammatory loop. That distinction matters because it changes where you intervene. It shifts the target from “remove damaged tissue” to “correct or eliminate dysfunctional immune states.”
For longevity biotech, that’s not a semantic difference. It’s the difference between broadly suppressing inflammation and precisely dismantling one of its engines.
The Immune System’s Quiet Drift Into Dysfunction
Macrophages are, in many ways, the custodians of tissue health. They patrol, engulf debris, recycle lipids, and coordinate repair. Their flexibility is their strength. They can shift between inflammatory and reparative modes depending on context.
But that flexibility has a cost. Unlike short-lived immune cells, many macrophages persist for long periods, especially in tissues like the liver. Over time, they accumulate stress signals: DNA damage, metabolic overload, oxidative stress. The question has been whether these pressures push macrophages into true senescence or simply into exaggerated activation states.
Senescence itself is a specific condition. It involves stable cell cycle arrest, resistance to apoptosis, and the secretion of inflammatory factors known as the senescence-associated secretory phenotype (SASP). Traditionally, it’s been studied in fibroblasts and epithelial cells, not immune cells. Macrophages were assumed to behave differently, too dynamic to become fixed in place.
This paper challenges that assumption. It suggests that macrophages can indeed enter a stable, senescence-like state. Not transient, not reversible, but persistent and structurally distinct.
Recreating Aging in a Dish, Then Finding It in Tissues
To test whether macrophages can become truly senescent, the researchers didn’t rely on observational data alone. They forced the issue. Using ionizing radiation and chemotherapy agents, they induced DNA damage in macrophages and then waited.
The waiting is important. Acute stress responses can look like senescence, but they fade. What the researchers looked for instead was durability. Over the course of days, the macrophages stopped dividing, enlarged, lost key nuclear structural proteins, and developed biochemical hallmarks of senescence.
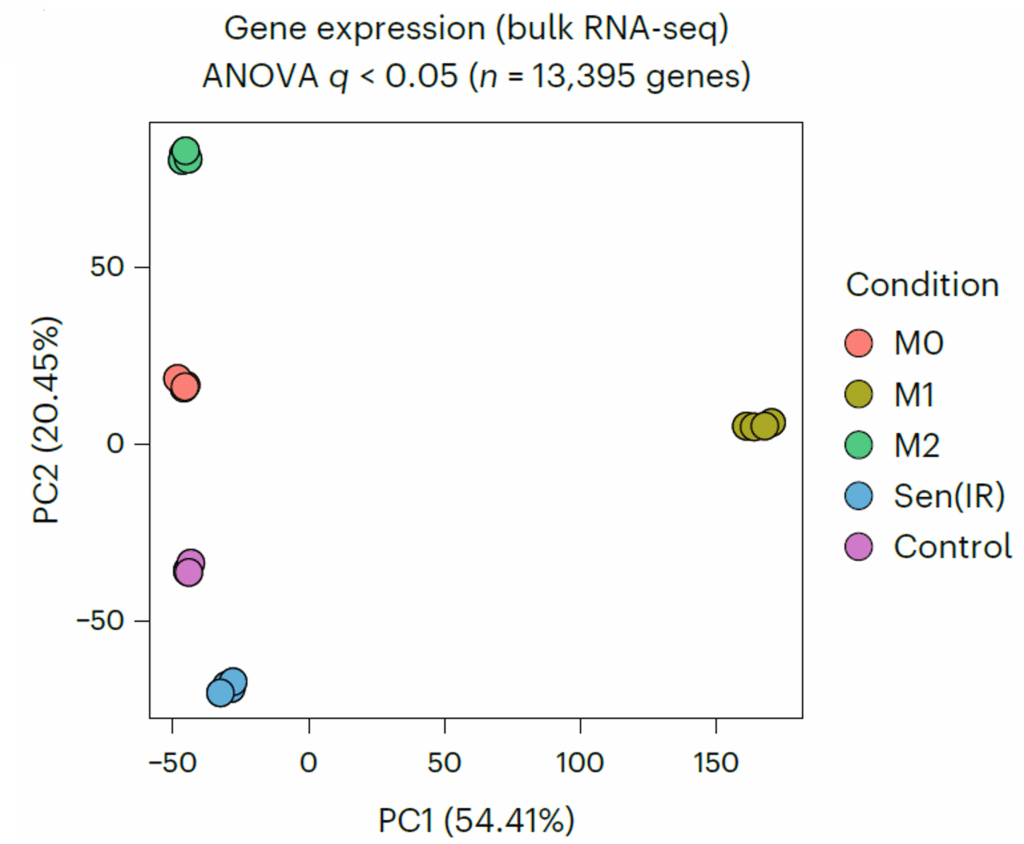
But the critical step was comparison. They placed these cells alongside classical macrophage states: M0, M1, and M2. These categories represent resting, inflammatory, and reparative macrophages, respectively. If senescent macrophages were just an extreme version of one of these, they would cluster with them.
They didn’t. Across RNA sequencing, proteomics, and metabolomics, the senescent macrophages formed their own distinct group. Not a subtype of activation, but a separate state altogether.
A New Identity: p21+TREM2+ Macrophages
The defining signature of these cells wasn’t just one marker. It was a combination. High levels of p21, a cell cycle inhibitor, paired with high levels of TREM2, a receptor associated with lipid handling and disease-associated macrophages.
This combination matters because it separates these cells from other immune states. Traditional markers like p16 didn’t track as cleanly, and the overlap with M1/M2 markers made simple classification unreliable. The p21+TREM2+ profile offered a clearer identity.
Mechanistically, these cells weren’t passive. They were metabolically active, but in a distorted way. They accumulated lipids, upregulated cholesterol metabolism pathways, and altered their handling of cellular debris. One of the more consequential changes was a reduction in efferocytosis, the process by which macrophages clear dead cells.
That failure creates a feedback loop. Dead cells accumulate, inflammation rises, and more macrophages are recruited into a dysfunctional environment. It’s less like a broken cleanup crew and more like a cleanup crew that starts generating its own mess.
The Signal That Won’t Turn Off
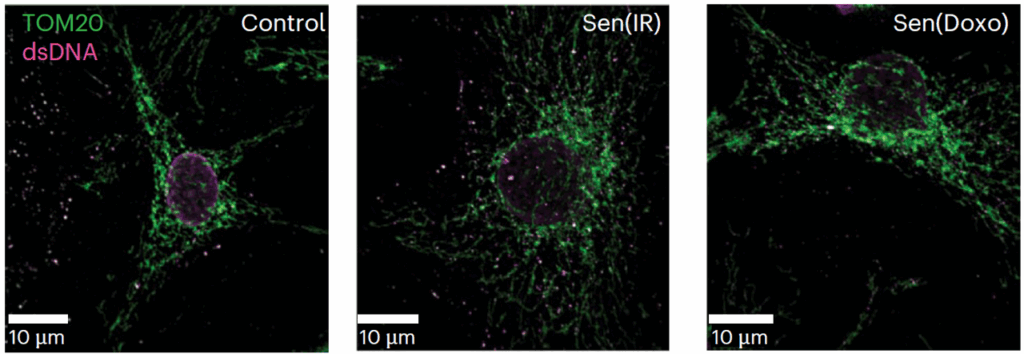
At the molecular level, the study points to a specific pathway: mitochondrial DNA leaking into the cytosol, activating the cGAS–STING pathway, and driving a type I interferon response.
This is a familiar pathway in antiviral defense. Cells use it to detect foreign DNA. But in this context, the DNA isn’t viral. It’s the cell’s own mitochondrial DNA, misplaced and misinterpreted.
That misinterpretation matters. It creates a chronic alarm signal, one that doesn’t resolve because the source of the signal persists. The macrophage becomes locked in a state of heightened sensitivity, responding aggressively to stimuli that would otherwise be benign.
The result is a sustained inflammatory tone. Not acute inflammation, which can be beneficial, but a low-grade, persistent signal. This is the essence of inflammaging, the chronic inflammation that accompanies aging and contributes to multiple diseases.
Aging Liver as a Case Study in Accumulation
The liver provided the clearest in vivo validation. In aged mice, macrophages known as Kupffer cells showed a dramatic increase in p21 expression. The proportion of senescent-like macrophages rose from a small minority to nearly half of the population.
That shift wasn’t just molecular. It correlated with functional decline. Lipid accumulation increased, inflammatory markers rose, and tissue structure deteriorated. The macrophages weren’t just present. They were participating.
Human data supported the pattern. In cirrhotic livers, macrophages carried a similar transcriptional signature. This doesn’t prove causality, but it strengthens the case that the phenomenon isn’t confined to mouse models.
The broader implication is that aging tissues may not simply accumulate damage. They may accumulate malfunctioning regulators of that damage, cells that should maintain homeostasis but instead disrupt it.
Clearing the Cells, Changing the Outcome
The most translationally relevant part of the study involved intervention. The researchers used ABT-263, a senolytic drug, to selectively eliminate senescent cells.
In vitro, it preferentially killed senescent macrophages over non-senescent ones. In vivo, it reduced the proportion of p21+ macrophages in the liver from roughly half to a small fraction. Alongside that reduction came measurable improvements: decreased inflammation, reduced lipid accumulation, and improved tissue appearance.
In a metabolic disease model, the effects extended further. Mice lost weight without eating less, inflammatory markers dropped, and gene expression shifted away from the senescent profile.
These results are compelling but not definitive. ABT-263 is not a precision tool. It affects multiple cell types, and its effects can’t be attributed solely to macrophage clearance. Still, the alignment between cell removal and physiological improvement suggests that these cells are not innocent bystanders.
A Refinement, Not a Revolution
This paper doesn’t overturn the concept of cellular senescence. It sharpens it. It suggests that senescence is not a uniform state but a collection of cell-type-specific programs, each with its own triggers and consequences.
For macrophages, the trigger appears to be a combination of DNA damage and lipid stress. The consequence is a hybrid state: part immune cell, part senescent cell, with features of both and the liabilities of each.
This refinement matters because it resolves a long-standing ambiguity. It provides a framework for distinguishing true senescence from activation. And it opens the door to more targeted interventions.
At the same time, it introduces new complexity. If senescence is heterogeneous, then so must be the therapies designed to address it.
Where the Evidence Still Falls Short
Despite the strength of the data, several uncertainties remain. The reliance on induced senescence models raises questions about how closely they mimic natural aging. The in vivo data are persuasive but still largely correlative.
There is also the issue of specificity. Macrophages are not a uniform population, and the p21+TREM2+ state may represent one of several dysfunctional trajectories. Whether this state dominates across tissues and conditions is still unclear.
Finally, translation to humans remains an open question. The human data show association, not intervention. And drugs like ABT-263 carry known risks, including hematological toxicity, which complicates their use in aging populations.
These are not minor caveats. They define the boundary between promising biology and viable therapy.
What This Means for Longevity
The broader implication is that aging may be driven as much by misregulated immune states as by damaged structural cells. That shifts attention toward immune modulation as a central strategy in longevity.
For senolytics, it suggests that part of their benefit may come from targeting immune compartments, not just fibroblasts or epithelial cells. For metabolic disease, it reinforces the idea that lipid handling and immune dysfunction are tightly linked.
For companies working on targeted senescence interventions, including vaccine-based approaches, the message is more nuanced. The target isn’t just “senescent cells.” It’s specific senescent cell states, defined by markers, pathways, and tissue context.
The field is moving from blunt instruments to more precise ones. This paper is a step in that direction, not the final word.
Source
Salladay-Perez, I. A., Avila, I., Estrada, L., Alexandru, A. C., Ponce, C., Dhingra, A., Torres, G., Deng, C. Y., Hegde, R., Gensheimer, J., Kale, A., Heckenbach, I., Hui, S., Edillor, C., Soto, J. A., Napior, A. J., Little, I., Larsen, M., Rose, J., … Covarrubias, A. J. (2026). . Nature Aging. Advance online publication. https://doi.org/10.1038/s43587-026-01101-6